python, 전처리, 통계, 가설검정, 기계학습, 회귀, 분류, 군집, 모델 학습, 모델 평가
평균 비교 검정(Mean Comparison Test)은 두 개 이상 집단 간 평균 차이가 단순한 우연인지 통계적으로 유의한지를 판단하는 방법이다. 범주형 변수로 집단을 나누고 연속형 변수의 평균을 비교한다. 이는 실무에서 가장 흔하게 사용되는 통계 기법 중 하나로, 약물 효과, 마케팅 전략, 제품 품질 등 다양한 분야에서 집단 간 차이를 과학적으로 검증하는 데 사용된다. 이 장에서는 t-검정과 ANOVA, 그리고 사후검정을 학습한다.
예제: 데이터 로드
import seaborn as snsimport pandas as pdimport numpy as npimport matplotlib.pyplot as pltfrom scipy import stats# 데이터 로드df = sns.load_dataset("penguins")print("데이터 크기:", df.shape)print("\n범주형 변수:", df.select_dtypes(include=['object', 'category']).columns.tolist())print("연속형 변수:", df.select_dtypes(include=[np.number]).columns.tolist())
평균 비교 검정은 집단 간 평균의 차이가 우연에 의한 것인지, 실제로 의미 있는 차이인지를 통계적으로 판단한다.
평균 비교 검정의 예시
질문
독립 변수 (범주형)
종속 변수 (연속형)
검정 방법
성별에 따라 몸무게가 다른가?
성별 (2그룹)
몸무게
독립표본 t-검정
종에 따라 부리 길이가 다른가?
종 (3그룹)
부리 길이
ANOVA
약물 투여 전후 혈압이 달라졌는가?
시점 (전/후)
혈압
대응표본 t-검정
교육 방법별 시험 점수가 다른가?
교육 방법 (4그룹)
시험 점수
ANOVA
15.1.1 검정 방법 선택 기준
평균 비교 검정 종류
상황
집단 수
표본 관계
사용 검정
조건
두 독립 집단 비교
2
독립
독립표본 t-검정
정규성, 등분산성
두 독립 집단 (등분산 X)
2
독립
Welch t-검정
정규성
동일 대상 전후 비교
2
대응
대응표본 t-검정
차이값의 정규성
세 집단 이상 비교
3+
독립
일원분산분석(ANOVA)
정규성, 등분산성
15.1.2 평균 비교 전 필수 가정 확인
평균 비교 검정은 다음 가정들을 전제로 한다.
검정 가정
가정
내용
확인 방법
위배 시 대응
1. 정규성
각 집단의 데이터가 정규분포를 따름
Shapiro-Wilk, Q-Q plot
비모수 검정, 변환
2. 등분산성
집단 간 분산이 동일함
Levene 검정
Welch 검정, Games-Howell
3. 독립성
각 관측치가 서로 독립적
실험 설계 확인
혼합 모델, 반복측정 ANOVA
가정 위배 시 강건성
표본 크기가 충분히 크면(n ≥ 30) 중심극한정리에 의해 정규성 가정이 완화됨
등분산성은 표본 크기가 집단 간 유사하면 어느 정도 강건함
독립성은 절대 위배되어서는 안 되는 가정
15.2 두 집단 평균 비교: 독립표본 t-검정
독립표본 t-검정(Independent Samples t-test)은 서로 독립된 두 집단의 평균을 비교하는 가장 기본적인 검정이다.
가설 설정
H₀ (귀무가설): μ₁ = μ₂ (두 집단의 모평균이 같다)
H₁ (대립가설): μ₁ ≠ μ₂ (두 집단의 모평균이 다르다)
t-통계량
\[
t = \frac{\bar{x}_1 - \bar{x}_2}{s_p \sqrt{\frac{1}{n_1} + \frac{1}{n_2}}}
\]
여기서 \(s_p\)는 합동 표준편차(pooled standard deviation)이다.
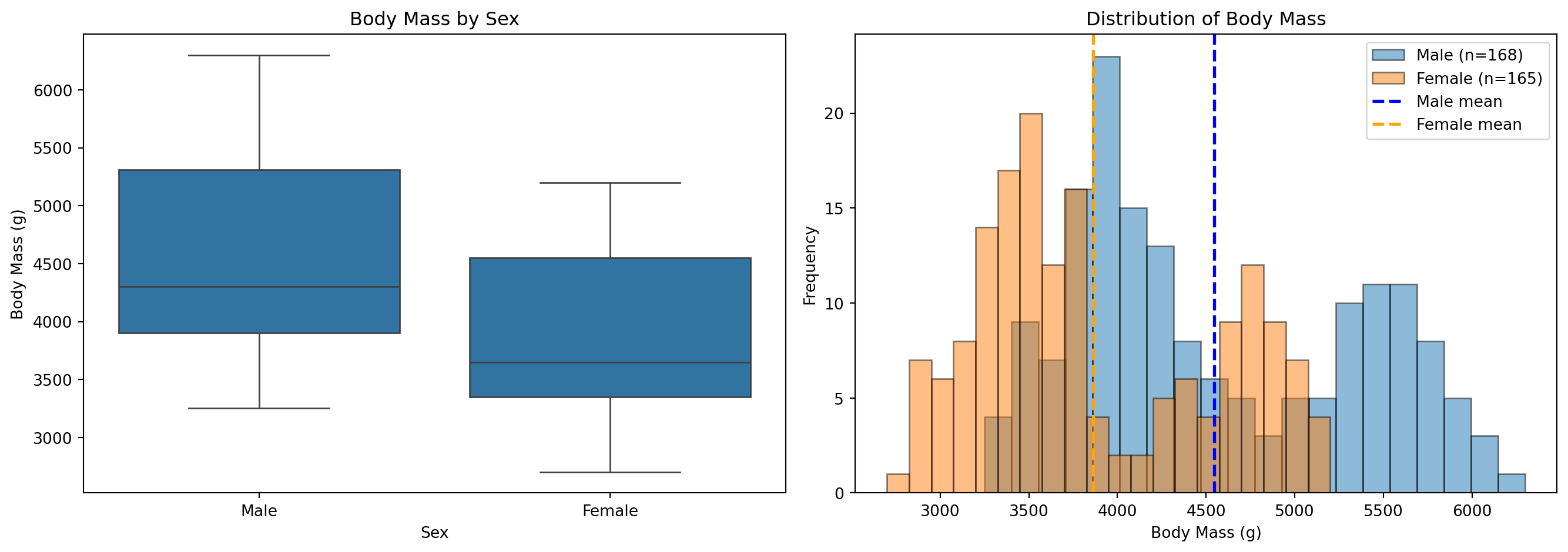
15.2.1 예제: 성별에 따른 체중 비교
예제: 데이터 준비 및 탐색
from scipy.stats import ttest_ind# 필요한 열만 선택 및 결측치 제거df_t = df[["sex", "body_mass_g"]].dropna()print("=== 집단별 기술 통계량 ===")summary = df_t.groupby("sex")["body_mass_g"].agg(['count', 'mean', 'std', 'min', 'max'])print(summary)# 집단 분리male = df_t[df_t["sex"] =="Male"]["body_mass_g"]female = df_t[df_t["sex"] =="Female"]["body_mass_g"]print(f"\n남성 표본 크기: {len(male)}")print(f"여성 표본 크기: {len(female)}")
=== 집단별 기술 통계량 ===
count mean std min max
sex
Female 165 3862.272727 666.172050 2700.0 5200.0
Male 168 4545.684524 787.628884 3250.0 6300.0
남성 표본 크기: 168
여성 표본 크기: 165
등분산 가정이 의심되면 Welch t-검정을 사용한다. 이는 등분산을 가정하지 않는 t-검정으로, 더 보수적인 결과를 제공한다.
예제: Welch t-검정
# Welch t-검정 (등분산 가정 불필요)t_stat_welch, p_value_welch = ttest_ind(male, female, equal_var=False)print("\n=== Welch t-검정 ===")print(f"t-통계량: {t_stat_welch:.4f}")print(f"p-value: {p_value_welch:.4f}")# Student vs Welch 비교print("\n=== t-검정 비교 ===")print(f"Student t-test: t = {t_stat:.4f}, p = {p_value:.4f}")print(f"Welch t-test: t = {t_stat_welch:.4f}, p = {p_value_welch:.4f}")
=== Welch t-검정 ===
t-통계량: 8.5545
p-value: 0.0000
=== t-검정 비교 ===
Student t-test: t = 8.5417, p = 0.0000
Welch t-test: t = 8.5545, p = 0.0000
Student vs Welch 선택 기준
등분산성 만족 → Student t-검정 (검정력 높음)
등분산성 의심 → Welch t-검정 (안전)
확신 없음 → Welch t-검정 (기본 선택 권장)
15.4 세 집단 이상 비교: 일원분산분석 (ANOVA)
일원분산분석(One-way ANOVA)은 세 개 이상 집단의 평균을 동시에 비교한다. 여러 번의 t-검정 대신 ANOVA를 사용하는 이유는 다중 비교로 인한 1종 오류 증가를 방지하기 위해서다.
가설 설정
H₀ (귀무가설): μ₁ = μ₂ = μ₃ = ⋯ = μₖ (모든 집단의 모평균이 같다)
H₁ (대립가설): 적어도 하나의 모평균이 다르다
F-통계량
\[
F = \frac{\text{집단 간 분산}}{\text{집단 내 분산}} = \frac{MSB}{MSW}
\]
F 값이 클수록 집단 간 차이가 집단 내 변동에 비해 크다는 의미이다.
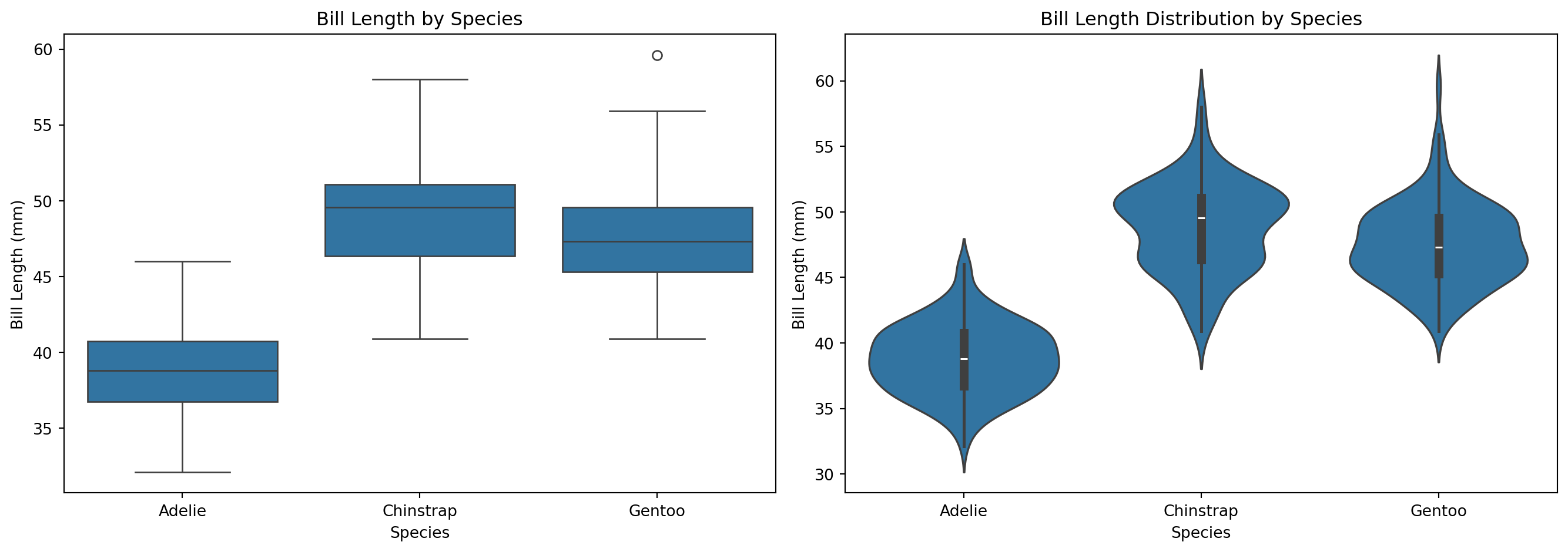
15.4.1 예제: 종별 부리 길이 비교
예제: 데이터 준비 및 탐색
from scipy.stats import f_oneway# 필요한 열 선택 및 결측치 제거df_a = df[["species", "bill_length_mm"]].dropna()print("=== 종별 부리 길이 기술 통계량 ===")summary = df_a.groupby("species")["bill_length_mm"].agg(['count', 'mean', 'std'])print(summary)# 집단별 데이터 분리adelie = df_a[df_a["species"] =="Adelie"]["bill_length_mm"]chinstrap = df_a[df_a["species"] =="Chinstrap"]["bill_length_mm"]gentoo = df_a[df_a["species"] =="Gentoo"]["bill_length_mm"]
=== 종별 부리 길이 기술 통계량 ===
count mean std
species
Adelie 151 38.791391 2.663405
Chinstrap 68 48.833824 3.339256
Gentoo 123 47.504878 3.081857
예제: 가정 확인
# 1. 각 집단의 정규성 검정print("\n=== 정규성 검정 ===")for species, data in [("Adelie", adelie), ("Chinstrap", chinstrap), ("Gentoo", gentoo)]: _, p = shapiro(data)print(f"{species}: p = {p:.4f}{'(정규)'if p >0.05else'(비정규)'}")# 2. 등분산성 검정print("\n=== 등분산성 검정 (Levene) ===")_, p_levene = levene(adelie, chinstrap, gentoo)print(f"p = {p_levene:.4f}{'(등분산)'if p_levene >0.05else'(이분산)'}")
=== 정규성 검정 ===
Adelie: p = 0.7166 (정규)
Chinstrap: p = 0.1941 (정규)
Gentoo: p = 0.0135 (비정규)
=== 등분산성 검정 (Levene) ===
p = 0.1078 (등분산)
예제: 일원분산분석
# ANOVA 수행f_stat, p_value = f_oneway(adelie, chinstrap, gentoo)print("\n=== 일원분산분석 (One-way ANOVA) ===")print(f"F-통계량: {f_stat:.4f}")print(f"p-value: {p_value:.4f}")# 결과 해석alpha =0.05print(f"\n유의수준 {alpha} 기준:")if p_value < alpha:print(f"✓ 귀무가설 기각: 종에 따른 부리 길이 차이가 유의함")print(f" → 사후검정(Post-hoc test) 필요")else:print(f"✗ 귀무가설 채택: 종에 따른 부리 길이 차이가 유의하지 않음")
=== 일원분산분석 (One-way ANOVA) ===
F-통계량: 410.6003
p-value: 0.0000
유의수준 0.05 기준:
✓ 귀무가설 기각: 종에 따른 부리 길이 차이가 유의함
→ 사후검정(Post-hoc test) 필요
예제: 시각화
# 시각화fig, axes = plt.subplots(1, 2, figsize=(14, 5))# 박스플롯sns.boxplot(x="species", y="bill_length_mm", data=df_a, ax=axes[0])axes[0].set_title("Bill Length by Species")axes[0].set_xlabel("Species")axes[0].set_ylabel("Bill Length (mm)")# 바이올린 플롯sns.violinplot(x="species", y="bill_length_mm", data=df_a, ax=axes[1])axes[1].set_title("Bill Length Distribution by Species")axes[1].set_xlabel("Species")axes[1].set_ylabel("Bill Length (mm)")plt.tight_layout()plt.show()
15.4.2 ANOVA의 한계와 사후검정 필요성
ANOVA는 “적어도 하나의 평균이 다르다”는 것만 알려주고, 구체적으로 어떤 집단 간 차이가 있는지는 알려주지 않는다. 이를 확인하기 위해 사후검정(Post-hoc Test)이 필요하다.
다중 비교 문제
여러 번의 t-검정을 반복하면 1종 오류(α)가 누적되어 증가한다.
3개 집단: 3번 비교 → 전체 α ≈ 0.14
4개 집단: 6번 비교 → 전체 α ≈ 0.26
5개 집단: 10번 비교 → 전체 α ≈ 0.40
사후검정은 이러한 다중 비교 문제를 보정한다.
15.5 대응표본 t-검정
대응표본 t-검정(Paired Samples t-test)은 동일한 대상의 전후 변화를 비교한다. 각 대상의 차이값(전 - 후)을 계산하고, 이 차이값의 평균이 0인지 검정한다.
가설 설정
H₀ (귀무가설): μd = 0 (전후 평균 차이가 0이다)
H₁ (대립가설): μd ≠ 0 (전후 평균 차이가 0이 아니다)
예제 구조 (개념적)
from scipy.stats import ttest_rel# 전후 데이터 (예시)# before = [120, 125, 130, 135, 128] # 처치 전 혈압# after = [115, 120, 125, 130, 122] # 처치 후 혈압# 대응표본 t-검정# t_stat, p_value = ttest_rel(before, after)# print(f"t-통계량: {t_stat:.4f}")# print(f"p-value: {p_value:.4f}")# # if p_value < 0.05:# print("처치 효과가 유의함")# else:# print("처치 효과가 유의하지 않음")
대응표본 vs 독립표본
구분
대응표본 t-검정
독립표본 t-검정
데이터 구조
동일 대상 전후
서로 다른 대상
예시
약물 투여 전후 혈압
남성 vs 여성 체중
장점
개인차 통제, 검정력 높음
구현 간단
가정
차이값의 정규성
각 집단의 정규성, 등분산성
15.6 사후검정 (Post-hoc Test)
ANOVA에서 유의한 결과가 나오면, 구체적으로 어떤 집단 간 차이가 있는지 확인하기 위해 사후검정을 수행한다.
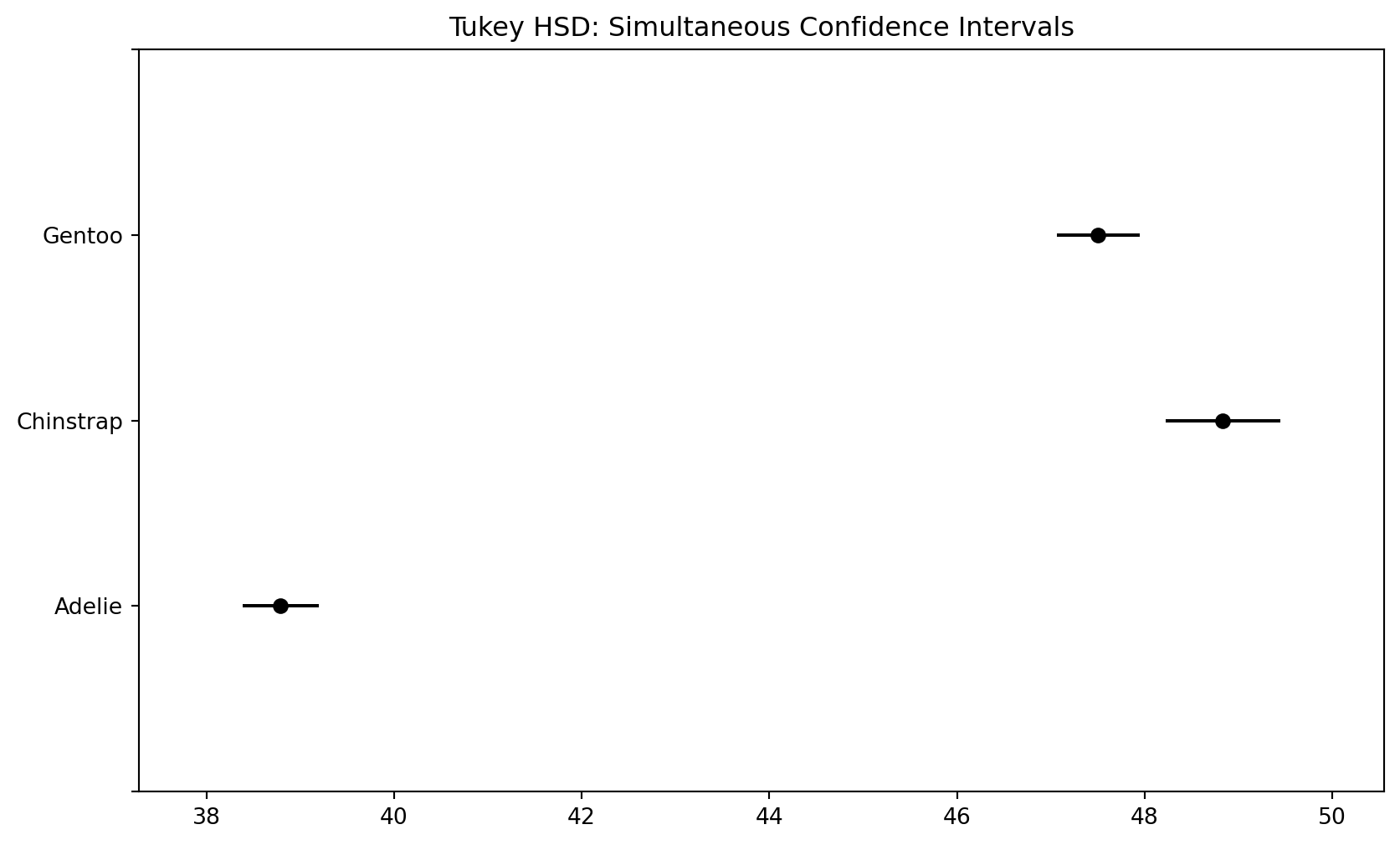
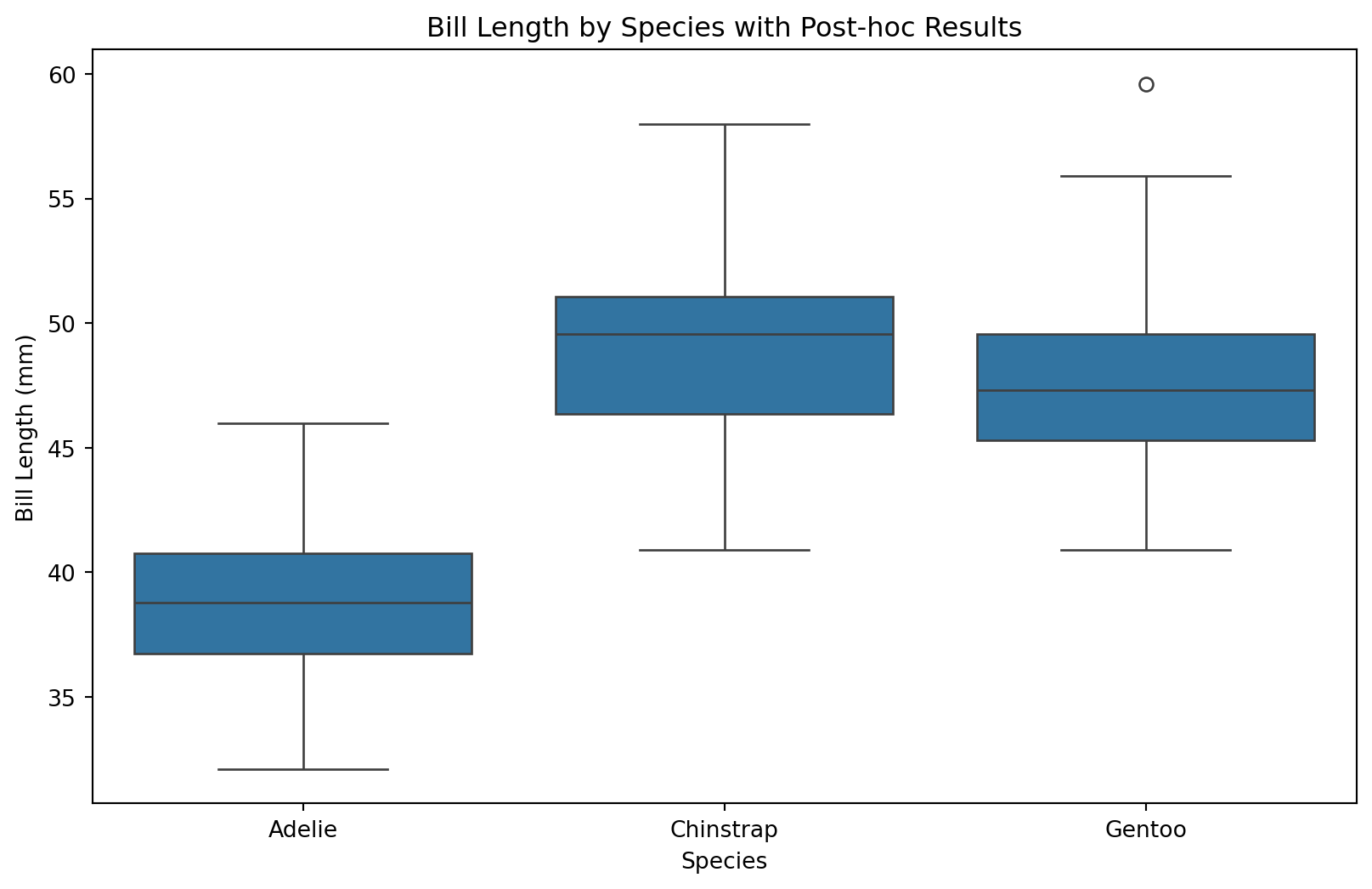
Tukey HSD는 ANOVA 이후 가장 표준적인 사후검정으로, 모든 집단 쌍의 평균 차이를 전체 유의수준을 유지하면서 비교한다.
예제: Tukey HSD 수행
from statsmodels.stats.multicomp import pairwise_tukeyhsd# Tukey HSD 사후검정tukey = pairwise_tukeyhsd( endog=df_a["bill_length_mm"], # 비교할 연속형 변수 groups=df_a["species"], # 집단 변수 alpha=0.05# 유의수준)print("=== Tukey HSD 사후검정 ===")print(tukey)
# Tukey HSD 결과 시각화tukey.plot_simultaneous()plt.title("Tukey HSD: Simultaneous Confidence Intervals")plt.show()# 박스플롯과 함께 비교plt.figure(figsize=(10, 6))sns.boxplot(x="species", y="bill_length_mm", data=df_a)plt.title("Bill Length by Species with Post-hoc Results")plt.xlabel("Species")plt.ylabel("Bill Length (mm)")# 유의한 차이가 있는 쌍에 별표 표시 (수동)# (실제로는 자동화된 시각화 패키지 사용 권장)plt.show()
15.6.3 Bonferroni 보정
Bonferroni 보정은 가장 보수적인 방법으로, 각 비교의 유의수준을 비교 횟수로 나눈다.